Open Data
Table of contents
At the SNaP Lab, we leverage multiple open access datasets to complete our research.
These datasets are hosted on Amarel. We share data storage with the Holmes lab (see Nodes and storage). All open access datasets are stored in /scratch/f_ah1491_1/open_data.
Below, we outline several datasets that have already undergone processing and are ready for use for certain analyses (mostly whole-brain connectivity analyses). Note, this document is a work in progress and will be imperfect. If you want to add content or make changes, please submit a pull request! (see Updating the wiki).
Datasets
Important
For each of the below datasets, please to do not write new data into any of the
derivativessubfolders without first discussing your planned analyses with Linden. Each of the subfolders insidederivativesis designed to store just a single set of outputs from a specific processing pipeline. For example, only files output by therun_mrtrix.shpiepline should be written to thederivatives/mrtrix_10msubdirectory. If you have specific processing needs, work with Linden to create a new pipeline (which needs to be stored in thescriptsdirectory) that will write outputs to its own subdirectory (e.g.,derivatives/your_pipeline).In general, once all subject-level data processing has been done, any further analyses should be carried out in your own project-specific folder (
/projectsp/f_lp756_1/[yourname]/[projectname]). See Nodes and storage for more details.
Human Connectome Project - Young Adult (HCP-YA)
Permissions
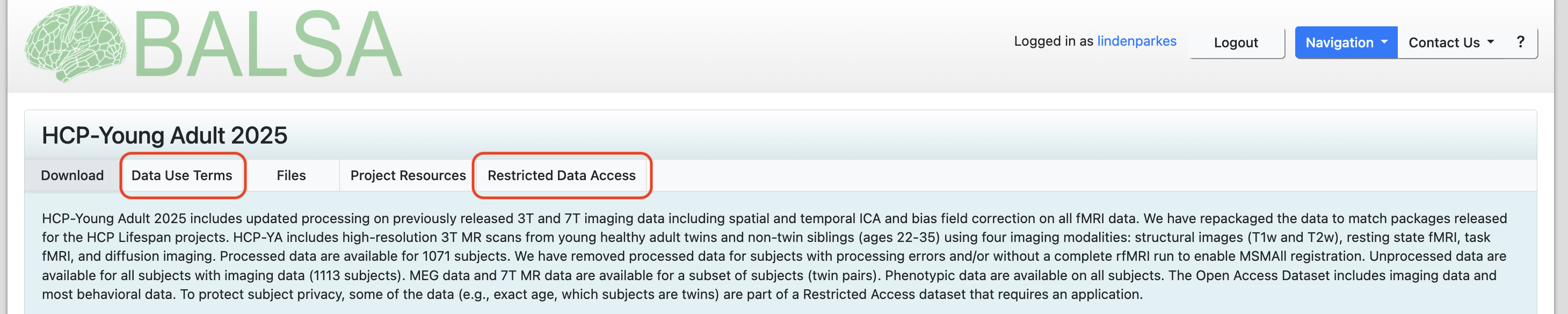
Before working with the HCP-YA, you will need to sign up for an account on BALSA. BALSA is where the raw and minimially preprocessed HCP data is stored. Depending on your project needs, you may not need to download any data from BALSA yourself. But, you will need to login to BALSA to accept the HCP-YA data use agreements. There are two data use agreements to read and sign, a standard one called Data Use Terms and a second one called Restricted Data Access. You will need to complete both before accessing the data on Amarel.
Relevant papers
Location
/scratch/f_ah1491_1/open_data/HCP_YA
File structure
HCP_YA
│ subject_ids.txt # list of subject IDs
│
└───HCP_1200 # minimally preprocessed data downloaded directly from HCP database
│ └───100206 # subject-level folder of downloaded data
│ │ └───...
│ └───...
│
└───collated_outputs # outputs from `derivatives/` wrapped up into easy-to-use .pkl files
│ │ hcpya_connectome.pkl # collated structural connectomes (from mrtrix_10m)
│ │ hcpya_myelin.pkl # collated myelin maps (from myelin_parcellated)
│ │ hcpya_rsfmri.pkl # collated resting-state fMRI time series (from fmri_parcellated)
│ │ hcpya_tfmri_contrasts.pkl # collated task-based fMRI contrasts (from fmri_parcellated_task-contrasts)
│ │ hcpya_tfmri.pkl # collated task-based fMRI time series (from fmri_parcellated)
│
└───derivatives # outputs from SNaP Lab processing pipelines
│ └───fmri_parcellated # parcellated fMRI data (rest and task)
│ │ └───100206
│ │ └───...
│ └───fmri_parcellated_task-contrasts # parcellated fMRI task-based contrasts (e.g., 2BK and 0BK)
│ │ └───100206
│ │ └───...
│ └───mrtrix_10m # whole-brain structural connectomes generated using MRTrix
│ │ └───100206
│ │ └───...
│ └───myelin_parcellated # parcellated myelin data (T1w/T2w ratio)
│ │ └───100206
│ │ └───...
│ └───parcellations # subject-specific parcellation files
│ │ └───100206
│ │ └───...
│
└───scripts # where scripts and pipelines are stored
│ │ download_data.py # downloads data into `HCP_1200`
│ │ run_parcgen.sh # generates subject-specific parcellation files (i.e., produces `derivatives/parcellations`)
│ │ run_parcellate_data.sh # parcellates fmri and myelin data (i.e., produces `derivatives/fmri_parcellated`, `derivatives/fmri_parcellated_task-contrasts`, and `derivatives/myelin_parcellated`)
│ │ run_mrtrix.sh # generates structural connectomes (i.e., produces `derivatives/mrtrix_10m`)
Microstructure-Informed Connectomics (MICA-MICs)
Permissions
The MICA-MICs permissions can be read and accepted here by following this link and clicking on the Terms of Use button.
Relevant papers
Location
/scratch/f_ah1491_1/open_data/MICA-MICs
File structure
MICA-MICs
│ subject_ids.txt # list of subject IDs
│
└───bids # raw data
│ └───sub-HC001 # subject-level folder of downloaded data
│ │ └───ses-01
│ │ │ └───anat # raw anatomical data (e.g., T1w)
│ │ │ └───dwi # raw diffusion data
│ │ │ └───fmap # raw fmap data
│ │ │ └───func # raw functional data
│ └───...
│ └───dataset_description.json
│
└───collated_outputs # outputs from `derivatives/` wrapped up into easy-to-use .pkl files
│ │ mics_connectome.pkl # collated structural connectomes (from mrtrix_10m)
│ │ mics_rsfmri.pkl # collated resting-state fMRI time series (from fmri_parcellated)
│
└───derivatives # outputs from SNaP Lab processing pipelines
│ └───fmriprep # fmriprep outputs
│ │ └───sub-HC001
│ │ │ sub-HC001.html
│ │ └───...
│ │ └───dataset_description.json
│ └───freesurfer # freesurfer outputs
│ │ └───sub-HC001
│ │ └───...
│ │ └───fsaverage
│ └───mrtrix_10m # whole-brain structural connectomes generated using MRTrix
│ │ └───sub-HC001
│ │ └───...
│ └───parcellations # subject-specific parcellation files
│ │ └───sub-HC001
│ │ └───...
│ └───xcpd # fmri postprocessing via xcp-d
│ │ └───sub-HC001
│ │ │ sub-HC001.html
│ │ └───...
│ │ └───dataset_description.json
│
└───scripts # where scripts and pipelines are stored
│ │ run_fmriprep.sh # generates outputs in `derivatives/fmriprep`
│ │ run_freesurfer.sh # generates outputs in `derivatives/freesurfer`
│ │ run_mrtrix.sh # generates outputs in `derivatives/mrtrix_10m`
│ │ run_parcgen.sh # generates outputs in `derivatives/parcellations`
│ │ run_xcpd.sh # generates outputs in `derivatives/xcpd`
│
└───MICs # processing outputs by the MICA team using Micapipe (see Cruces et al., 2022)
│ └───derivatives
│
└───download # data downloaded from OSF
About the collated_outputs
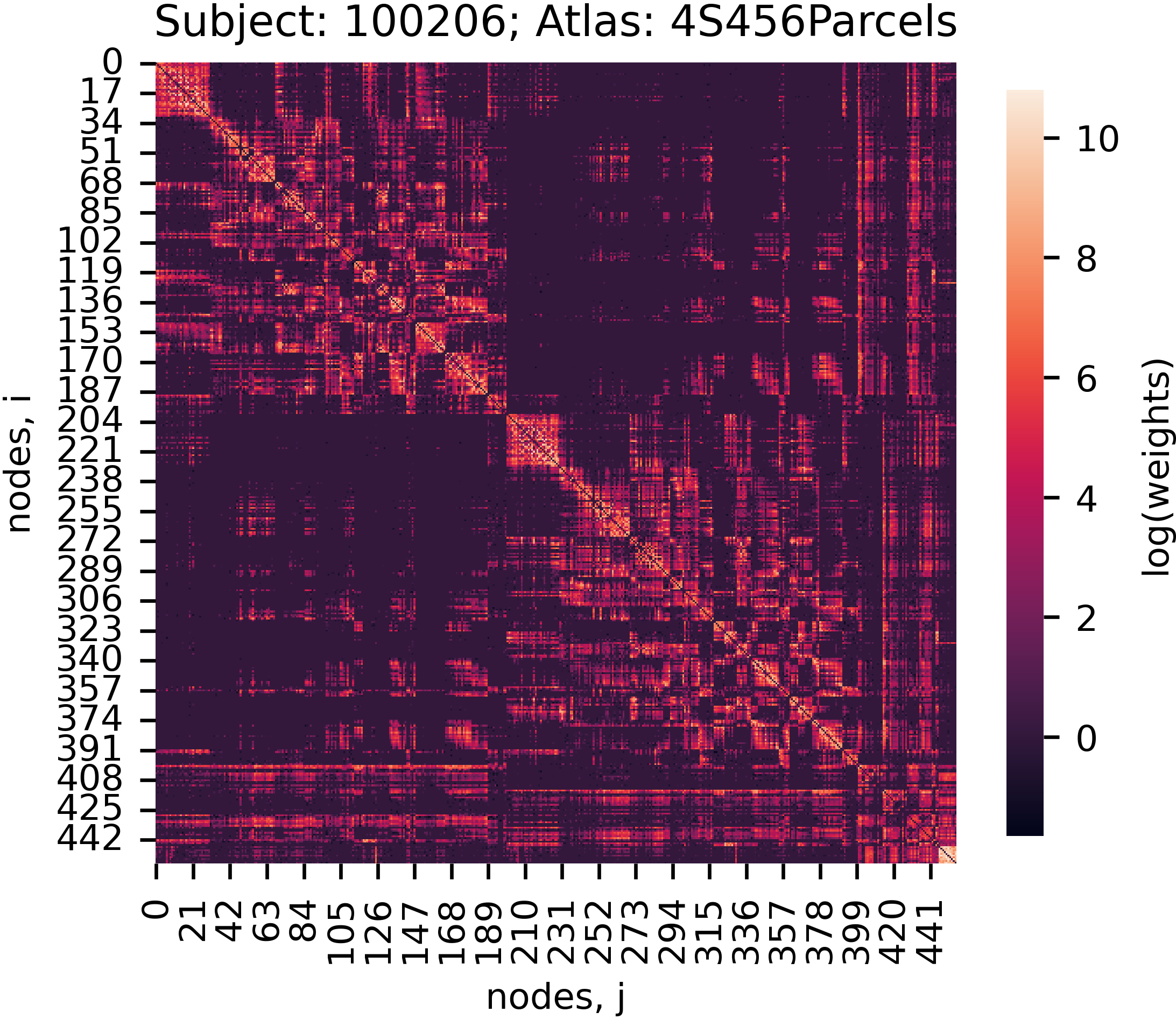
For most of the above datasets, you will find a subdirectory called collated_outputs inside the project folder. Inside collated_outputs, you will find a set of pickle files (.pkl) that store the derivatives from each pipeline in a single dictionary. These files are designed to facilitate quick and easy access to the outputs from our pipelines. If you want to get started with an analysis, downloading the collated_outputs onto your laptop is likely going to be the quickest way forward. See below example for how to work with these files in Python:
import os
import numpy as np
import seaborn as sns
# Location of collated outputs
in_dir = '/scratch/f_ah1491_1/open_data/HCP_YA/collated_outputs'
# Load pickle file. This will create a Python dictionary in your workspace where keys correspond to subject ids
with open(os.path.join(in_dir, 'hcpya_connectome.pkl'), 'rb') as handle:
data = pickle.load(handle)
print(list(data.keys())[:5])
# ['100206', '100307', '100408', '100610', '101006']
# Examine one subject. Data for each subject is also stored as a Python dictionary where, in this case, keys correspond to parcellations
print(data['100206'].keys())
# dict_keys(['Schaefer1007', 'Schaefer2007', 'Schaefer4007', '4S156Parcels', '4S256Parcels', '4S456Parcels'])
# Extract and plot a connectome
subject_id = '100206'
atlas = '4S456Parcels'
adjacency = data[subject_id][atlas]
adjacency_log = np.log(adjacency, out=np.zeros_like(adjacency), where=(adjacency != 0))
f, ax = plt.subplots(figsize=(4, 4))
sns.heatmap(adjacency_log, ax=ax, square=True, cbar_kws={'shrink': 0.75, 'label': 'log(weights)'})
ax.set_ylabel('nodes, i')
ax.set_xlabel('nodes, j')
ax.set_title('Subject: {0}; Atlas: {1}'.format(subject_id, atlas))
plt.show()